
„“(chuЁӨng)РВ”ө“ю·ЗТАЩҮРФІЙјҜУГУЪҸНлs»щЩ|ДҝҳЛө°°ЧЩ|өД¶ЁБҝ·ЦОц
ҸҲӮҘ1 Reiko Kiyonami2 ҪӯҚҳ*1 кҗӮҘ1
1(ЩҗД¬пwКА –ҝЖјј(ЦРҮш)УРПЮ№«Лҫ,ЙПәЈ201206) 2(ThermoFisher Scientific, San Jose, CA, USA)
ХӘТӘ ”ө“ю·ЗТАЩҮРФІЙјҜ(DIA)КЗлSЦш¶ЁБҝө°°ЧЩ|ҪMҢW¶шҪЁБўөДЩ|ЧV’ЯГијјРgЎЈDIA ДЬүт«@өГ’ЯГи·¶ҮъғИЛщУРДёлxЧУј°¶юјүЧУлxЧУРЕПў,І»•юФміЙөНШS¶ИлxЧУРЕПўөДҒGК§,Н¬•rН»ЖЖБЛёЯ·ЦұжЩ|ЧV¶юјү¶ЁБҝөДНЁБҝПЮЦЖЎЈұҫСРҫҝ»щУЪмoлҠҲцЬүөАЪеQ-qIT-OT ИэәПТ»Щ|ЧV,°l(fЁЎ)Х№БЛҪӣөдDIA ·Ҫ·ЁТФј°WiSIM-DIA әНFull MS-DIAғЙ·NИ«РВDIA ·Ҫ·Ё,ІўҢҰHela јҡ°ыИ«ө°°ЧЦРМнјУөД10 —lөНқв¶ИлД¶ОЯMРР¶ЁБҝ·ЦОц,ҝјІм·Ҫ·ЁөДҫҖРФЎўЦШ¬F(xiЁӨn)РФәНм`Гф¶ИЎЈҪY№ыұнГч,3 ·N·Ҫ·ЁөД¶ЁБҝПЮҫщөНЦБamol (14 ~435 amol),ІўХ№КҫіцБјәГөДҫҖРФәН¶ЁРФҙ_ЧCҝЙҝҝРФЎЈЖдЦР,WiSIM-DIA »щУЪі¬ёЯ·ЦұжТ»јүұO(jiЁЎn)ңy¶ЁБҝ,ЕcҪӣөдDIA ғһ(yЁӯu)„Э»ҘСa;Full MS-DIA өДЯx“сҙ°ҝЪғH3 amu,ДЬүтЦұҪУЯMРРЛСҺмиb¶Ё,ҢҚ¬F(xiЁӨn)БЛ”ө“юТАЩҮРФІЙјҜ(DDA)әНDIA өДҪy(tЁҜng)Т»,”[Г“БЛDIA ТАЩҮУЪDDA ҪЁБўЧVҲDҺмөДҫЦПЮРФЎЈ
кPжIФ~ мoлҠҲцЬүөАЪе; ”ө“ю·ЗТАЩҮРФІЙјҜ; ө°°ЧЩ|ҪMҢW; Ҫ^ҢҰ¶ЁБҝ
1ТэСФ
”ө“юТАЩҮРФІЙјҜ(Data dependent acquisition, DDA)КЗҙ®В“(liЁўn)Щ|ЧV·ЗДҝҳЛ»ҜәПОп·ЦОцөДЦчТӘКЦ¶ОЎЈө°°ЧЩ|ҪMҢWөДҪӣөдІЯВФ-шBҳҢ·Ё(Shotgun)јҙ»щУЪDDA °l(fЁЎ)Х№¶шҒн,АыУГТ»јүИ«’ЯГиҷzңyлД¶ОДёлxЧУ,И»әу°ҙРЕМ–ҸҠ¶ИЕЕБР,ҢўЗ°ИфёЙО»өДДёлxЧУТАҙОЯx“сЎўЛйБС,Іў’ЯГи¶юјүЛйЖ¬лxЧУЎЈН¬•r,„У‘B(tЁӨi)ЕЕіэЎўғr‘B(tЁӨi)ЕЕіэЎўЦРРФҒGК§/ Ф\”алxЧУУ|°l(fЁЎ)өИјјРg,К№DDA ұMҝЙДЬ¶аөШІЙјҜУРР§лД¶ОЧVҲD,ҢҚ¬F(xiЁӨn)иb¶ЁҪY№ыЧоҙу»Ҝ[1] ЎЈ»щУЪшBҳҢ·Ё,ө°°ЧЩ|ҪMҢWТСҪӣҢҚ¬F(xiЁӨn)ҪНДёө°°ЧЩ|ҪMҪУҪьНкИ«ёІЙw,ИЛоҗө°°ЧЩ|ҪMТІТСЯ_өҪ50% ТФЙПөД»щТтҪMёІЙwәН7 ӮҖ”өБҝјүөД„У‘B(tЁӨi)·¶Үъ[2,3] ЎЈИ»¶ш,DDA өДҫЦПЮРФТІЦрқuп@¬F(xiЁӨn):(1) ПИҸҠәуИхөДІЙјҜ·ҪКҪТЧФміЙөНШS¶ИлД¶ОРЕПўҒGК§;(2) ДёлxЧУЯx“сУРТ»¶ЁөДлSҷCРФ,ФміЙЦШ¬F(xiЁӨn)РФІ»јС;(3) ГҝӮҖСӯӯh(huЁўn)«@өГөДЧVҲD”өБҝІ»Т»,ФміЙ’ЯГиьc”өІ»ҫщ„т,Ӱ푶ЁБҝ·ЦОцңКҙ_РФЎЈ
ДҝҳЛө°°ЧЩ|ҪMҢWбҳҢҰДҝҳЛө°°Ч/ лД¶ОлxЧУҢҚ•rұO(jiЁЎn)ңyәНІЙјҜ,ұЬГвБЛDDA өДРЕПўҒGК§әНЦШ¬F(xiЁӨn)РФҶ–о}ЎЈЦчТӘІЙјҜ·Ҫ·Ё°ьАЁЯx“слxЧУұO(jiЁЎn)ңy(Selected ion monitoring, SIM)Ўў»щУЪИэЦШЛДҳO—UөДЯx“с·ҙ‘ӘұO(jiЁЎn)ңy(Selected reaction monitoring, SRM)әН»щУЪёЯ·ЦұжЩ|ЧVөДЖҪРР·ҙ‘ӘұO(jiЁЎn)ңy(Parallel reaction monitoring, PRM),КЗДҝҳЛө°°ЧтһЧCәНҪ^ҢҰ¶ЁБҝөДУРР§КЦ¶О[4,5] ЎЈө«КЗДҝҳЛРФөДІЙјҜ·Ҫ·ЁРиТӘЦё¶ЁДҝҳЛлД¶О,ҢҰУЪОҙЦӘлД¶Оҹo·ЁІЙјҜ;НЁБҝПЮЦЖТІК№өГТ»ҙОҢҚтһЦ»ДЬұO(jiЁЎn)ңy”өБҝУРПЮлД¶О»тлxЧУҢҰ,лyТФқMЧгҙуТҺ(guЁ©)ДЈө°°Ч·ЦОцөДРиТӘЎЈ
”ө“ю·ЗТАЩҮРФІЙјҜ(Data independent acquisition, DIA)К№УГ25 amu »тёьҙуйgёфҢўХыӮҖЩ|Бҝ·¶ҮъөИ·ЦһйИфёЙҙ°ҝЪ,ГҝӮҖҙ°ҝЪТАҙОЯx“сЎўЛйБСЎў’ЯГиЎЈDIA ДЬүт«@өГЩ|Бҝ·¶ҮъғИЛщУРДёлxЧУөДИ«ІҝЛйЖ¬лxЧУРЕПў,НЁБҝҹoЙППЮ,Сӯӯh(huЁўn)•rйg№М¶Ё,Н¬•r”ө“юҝЙТФ»ШЛЭ,УРР§ҪвӣQБЛDDA әНДҝҳЛІЙјҜ·Ҫ·ЁҙжФЪөДҶ–о}[6] ЎЈДҝЗ°,ТС°l(fЁЎ)Х№БЛ¶а·N»щУЪпwРР•rйg(Q-TOF)ЎўмoлҠҲцЬүөАЪе(Orbitrap)әНлxЧУЪеөДDIA ·Ҫ·Ё[7] ЎЈGillet өИК№УГ32 ӮҖЯBАm(xЁҙ)өД25 amu ҙ°ҝЪ,»щУЪQ-TOF (TripleTOF 5600)°l(fЁЎ)Х№БЛSWATH јјРg,ІўЧCГчФ“јјРgөД¶ЁБҝДЬБҰЕcSRM Па®”[8] ЎЈEgertson өИАыУГQ-Orbitrap (Q Exactive)ӘҡУРөД¶аЦШ’ЯГи№ҰДЬ(Multiplexing,MSX)°l(fЁЎ)Х№БЛMSX-DIA јјРg,ҢўЯx“сҙ°ҝЪҝsРЎөҪ4 amu,ЧоҙуіМ¶ИңpЙЩБЛ№ІБчіцлД¶ОәНлsЩ|өДёЙ”_[9] ЎЈ
И»¶ш,ӮчҪy(tЁҜng)”ө“ю·ЗТАЩҮРФІЙјҜИФҙжФЪЦT¶аҫЦПЮ:(1) УЙУЪ’ЯГиЛЩ¶ИөДПЮЦЖ,DIA лyТФК№УГі¬ёЯ·ЦұжВК’ЯГи;(2) DIA өДҙуҙ°ҝЪЯx“сТэИлЭ^ҙуёЙ”_,лmИ»MSX-DIA ҝsРЎБЛЯx“сҙ°ҝЪ,ө«РиТӘМШ¶ЁөДЛг·ЁҪвОц”ө“ю,ФцјУБЛ№ӨЧчБҝ;(3) DIA ТАЩҮУЪDDA ҪЁБўөДЧVҲDҺмЯMРРЖҘЕд,ҢҚ¬F(xiЁӨn)¶ЁРФҙ_ЧCәН¶ЁБҝлxЧУЯx“с,ТтҙЛDDA иb¶ЁІ»өҪөДө°°Ч,DIA ТІҹo·Ё·ЦОцЎЈ
ұҫСРҫҝ»щУЪЛДҳO—U-мoлҠҲцЬүөАЪе-ҫҖРФлxЧУЪе(Q-OT-qIT)ИэәПТ»Щ|ЧV,АыУГМнјУ10 —lөНқв¶ИлД¶ОөДHela ҳУұҫ,°l(fЁЎ)Х№ІўҝјІмБЛ3 ·N”ө“ю·ЗТАЩҮРФІЙјҜ·Ҫ·Ё,°ьАЁҪӣөдөДDIAЎўИ«РВөДҢ’ҙ°ҝЪSIM ’ЯГиDIA(WiSIM-DIA)әНИ«’ЯГиDIA (Full MS-DIA),¶ЁБҝПЮҫщЯ_өҪamol Л®ЖҪ,ҫҖРФЎўЦШ¬F(xiЁӨn)РФБјәГЎЈЖдЦР,WiSIM-DIA әНFull MS-DIA »щУЪ24 Иfі¬ёЯ·ЦұжВК,АыУГТ»јүҫ«ҙ_Щ|Бҝ”ө¶ЁБҝЎў¶юјүлxЧУЪе¶ЁРФҙ_ЧC,ЯMТ»ІҪҝsРЎБЛЯx“сҙ°ҝЪ,МбёЯБЛҷzңyМШ®җРФЎЈҙЛНв,Full-MS DIA ҝЙТФЦұҪУЛСҺм,ҢҚ¬F(xiЁӨn)БЛDDA ЕcDIA өДҪy(tЁҜng)Т»,ө°°Чиb¶Ё”өБҝЕcDDA Па®”,”[Г“БЛЧVҲDҺмөДПЮЦЖЎЈ»щУЪQ-OT-qIT өД”ө“ю·ЗТАЩҮРФІЙјҜ·Ҫ·Ём`»о¶аҳУЎўБчіМәҶҶОУРР§,ФЪДҝҳЛө°°ЧЩ|ҪMҢWоIУтҫЯУРҸVйҹөД‘ӘУГЗ°ҫ°ЎЈ
2ҢҚтһІҝ·Ц
2.1 ғxЖчЕcФҮ„©
Orbitrap Fusion ИэәПТ»Щ|ЧVғxЎўEASY-nLC 1000 ј{Бчі¬ёЯР§ТәПаЙ«ЧV(Thermo Fisher Scientific)ЎЈҳЛңКлД¶ОУЙЙъ№ӨЙъОп(ЙПәЈ)№Й·ЭУРПЮ№«ЛҫәПіЙ;ТТлжЎўјЧЛб(Thermo Fisher Scientific);ЖдЛьФҮ„©ҫщЩҸЧФSigma-Aldrich №«ЛҫЎЈ
2. 2Hela јҡ°ыИ«ө°°ЧГёҪвТәЦЖӮд
ИЎЯmБҝHela јҡ°ыіБөн,јУИлә¬7 mol/ L ДтЛШЎў2 mol/ L БтлеЎў1 mmol/ L ұҪјЧ»щ»ЗхЈ·ъ(PMSF) әН50 mmol/ L ¶юБтМKМЗҙј(DTT)өДө°°ЧМбИЎТә,ФЪі¬В•јҡ°ыЖЖЛйғxЦРі¬В•3 ҙОёч5 sЎЈұщЙП·ЕЦГ20 minәу,4 ТжлxРД(15000 g) 30 min, ИЎЙПЗеТә,Bradford ·Ёҙ_¶ЁҝӮө°°Чқв¶ИЎЈПтө°°ЧМбИЎТәЦРјУИлDTT(ҪKқв¶И10 mmol/ L),УЪ56 ТжХсКҺ30 minЎЈјУИлөвТТхЈ°·(ҪKқв¶И50 mmol/ L),КТңШұЬ№вХсКҺ40 minЎЈ·ҙ‘ӘәујУИл6 ұ¶уw·eөДоAАдұыНӘ,-20 Тж·ЕЦГ3 h,4 ТжлxРД(15000 g)30 min, —үЙПЗеТәЎЈҢўө°°ЧіБөнИЬУЪ50 mmol/ L NH4HCO3 ИЬТә,јУИлТИө°°ЧГёTrypsin(ГёоUө°°Чһй1оU40, w / w),37 ТжГёҪвЯ^Т№ЎЈҢўИ«ө°°ЧГёҪвТәПЎбҢЦБҪKқв¶Иһй100 mg/ LЎЈ
2.3 ҳЛңКлД¶ОМнјУ
өИЦШ·QИЎ10 —lҳЛңКлД¶О·ЫД©,»мәПәуК№УГ0. 1%јЧЛбИЬТәід·ЦИЬҪвһй1 g/ L лД¶О»мәПИЬТә(ҝӮқв¶И10 g/ L)ЎЈлД¶О»мәПИЬТәЯMТ»ІҪК№УГ100 mg/ L Hela ГёҪвТәЦрјүПЎбҢһй7 ·Nқв¶ИМЭ¶И(200, 50, 10, 2, 0.5,0.1 әН0.02 mg/ L)ИЬТәЎЈБнИЎЙЩБҝҳЛңКлД¶ОИЬТәУГЛ®ПЎбҢЦБ500 mg/ L, УГЧчPRM ’ЯГи,ҳӢҪЁЧVҲDҺмЎЈ
2.4 ј{БчТәПаЙ«ЧV·Ҫ·Ё
Й«ЧVЦщ:ЧФЦЖј{БчC18 Й«ЧVЦщ(3 ЧМm, 100 фҶ, 75 ЧМm ТБ15 cm);ЙПҳУБҝ: 1 ЧМL;БчЛЩ:300 nL/ min;A Па: 0. 1%јЧЛбИЬТә;B Па: 0. 1% јЧЛб-ТТлжИЬТә;·ЦОцМЭ¶И:0 ~4 min,3% ~10% B;4 ~74 min,10%~35% B;74 ~78 min,35% ~90% B;78 ~90 min,90% BЎЈ
2.5 Щ|ЧV·Ҫ·Ё
лxЧУФҙ:nano-Flex ј{ҮҠмFлxЧУФҙ;’ЯГиДЈКҪ:ХэлxЧУ;ҮҠмFлҠүә:2. 2 kV;лxЧУӮчЭ”№ЬңШ¶И:275 Тж;RF-lens:60%ЎЈ
DDA ’ЯГи:Т»јү’ЯГиДЈКҪ:И«’ЯГи;Т»јү’ЯГи·¶Үъ:m / z 300 ~ 1800;Т»јүҷzңy:Orbitrap(·ЦұжВК240 K);Яx“сДЈКҪ:ЛДҳO—U;Яx“сҙ°ҝЪ:2 amu;ЛйБСДЈКҪ:CID;ЛйБСДЬБҝ:30%;¶юјүҷzңy:лxЧУЪе;„У‘B(tЁӨi)ЕЕіэ:60 s;Сӯӯh(huЁўn)•rйg:3 sЎЈ
PRM ’ЯГи:ДёлxЧУБРұн:10 —lҳЛңКлД¶ОөДҫ«ҙ_Щ|Бҝ”ө;Яx“сДЈКҪ:ЛДҳO—U;Яx“сҙ°ҝЪ:2 amu;ЛйБСДЈКҪ:HCD;ЛйБСДЬБҝ:30%;¶юјүҷzңy:Orbitrap(·ЦұжВК30 K);Сӯӯh(huЁўn)ДЈКҪ:°ҙДёлxЧУБРұнТАҙО’ЯГиЎЈ
DIA ·Ҫ·Ё:ДёлxЧУБРұн:m / z 510 ~890, йgёф20 amu;Яx“сДЈКҪ:ЛДҳO—U;Яx“сҙ°ҝЪ:20 amu;ЛйБСДЈКҪ:HCD;ЛйБСДЬБҝ:30%;¶юјүҷzңy:Orbitrap(·ЦұжВК30 K);¶юјү’ЯГи·¶Үъ:m / z 150 ~2000;Сӯӯh(huЁўn)ДЈКҪ:°ҙДёлxЧУБРұнТАҙО’ЯГиЎЈ
WiSIM-DIA ·Ҫ·Ё:Т»јү’ЯГиДЈКҪ:SIM;Т»јүЯx“сДЈКҪ:лxЧУЪе;Т»јү’ЯГи·¶Үъ:m / z 400 ~ 600, m / z600 ~800, m / z 800 ~1000;Т»јүҷzңy:Orbitrap(·ЦұжВК240 K);ДёлxЧУБРұн:m / z 406 ~598, m / z 606 ~798, m / z 806 ~998 йgёф12 amu;¶юјүЯx“сДЈКҪ:ЛДҳO—U;Яx“сҙ°ҝЪ:12 amu;ЛйБСДЈКҪ:CID;ЛйБСДЬБҝ: 30%;¶юјүҷzңy:лxЧУЪе;¶юјү’ЯГи·¶Үъ:m / z 150 ~2000;Сӯӯh(huЁўn)ДЈКҪ:3 ӮҖSIM+3 ӮҖДёлxЧУБРұнТАҙО’ЯГиЎЈFull MS-DIA ·Ҫ·Ё:Т»јү’ЯГиДЈКҪ:И«’ЯГи;Т»јү’ЯГи·¶Үъ: m / z 400 ~1000;Т»јүҷzңy:Orbitrap(·ЦұжВК240 K);ДёлxЧУБРұн:m / z 401. 5 ~518. 5, m / z 521. 5 ~638.5, m / z 641. 5 ~758. 5, m / z 761. 5 ~878. 5,m / z 881.5 ~998.5, йgёф3 amu;Яx“сДЈКҪ:ЛДҳO—U;Яx“сҙ°ҝЪ:3 amu;ЛйБСДЈКҪ:CID;ЛйБСДЬБҝ:30%;¶юјүҷzңy:лxЧУЪе;¶юјү’ЯГи·¶Үъ:m / z 150 ~2000;Сӯӯh(huЁўn)ДЈКҪ:5 ӮҖТ»јүИ«’ЯГи+5 ӮҖДёлxЧУБРұнТАҙО’ЯГиЎЈ
2.6 ”ө“юМҺАн
ҳЛңКлД¶ОPRM ”ө“юК№УГProteome Discoverer 1. 4 ЬӣјюЯMРРЛСҺмиb¶Ё,ДёлxЧУЩ|Бҝҫ«¶И:10 ppm,ЧУлxЧУЩ|Бҝҫ«¶И:0. 02 DaЎЈиb¶ЁҪY№ыЧчһйЧVҲDҺмЯMРРDIA өД¶ЁРФҙ_ЧCәН¶ЁБҝлxЧУМфЯxЎЈDIA ”ө“юК№УГPinpoint 1. 4 ЬӣјюЯMРРЧVҲDҺмлxЧУәYЯxЎў¶ЁРФҙ_ЧCЎў¶ЁБҝ·ЦОцәНҳЛңКЗъҫҖАLЦЖЎЈЩ|Бҝҫ«¶ИФOЦГН¬ЙП,ДёлxЧУ¶ЁБҝЯx“сҶОН¬О»ЛШ·еәНөЪТ»ӮҖН¬О»ЛШ·е,ЧУлxЧУ¶ЁБҝЯx“сҸҠ¶ИЧоёЯөД2 ӮҖy лxЧУ,¶ЁРФҙ_ЧCЯx“сҸҠ¶ИЧоёЯөД8 ӮҖЧУлxЧУЎЈ100 ng Hela јҡ°ыИ«ө°°ЧDDA әНFull MS-DIA ”ө“юК№УГProteome Discoverer 1. 4 ЬӣјюЯMРРUniprot ИЛоҗө°°Ч”ө“юҺмЛСҺмиb¶ЁЕcұИЭ^,Щ|Бҝҫ«¶ИФOЦГН¬ЙП(Full MS-DIA ДёлxЧУЩ|Бҝҫ«¶ИФOЦГһй1. 5 Da),ҝЙЧғРЮп—:јЧБт°ұЛбСх»Ҝ(M+15. 995 Da),№М¶ЁРЮп—:°ллЧ°ұЛблејЧ»щ»Ҝ(C+57. 021 Da)ЎЈиb¶ЁҪY№ыК№УГPercolator ЯMРРҮАёсҝЁЦө(q<0. 01)ЎЈ
3ҪY№ыЕcУ‘Х“
3.1 »щУЪQ-qIT-OT өД„“(chuЁӨng)РВ”ө“ю·ЗТАЩҮРФІЙјҜІЯВФөДҪЁБў
”ө“ю·ЗТАЩҮРФІЙјҜТАҙО’ЯГиХыӮҖЩ|Бҝ·¶ҮъөДЛщУРДёлxЧУј°¶юјүЧУлxЧУ,Сӯӯh(huЁўn)•rйgйL,ТтҙЛҢҰЩ|ЧV’ЯГиЛЩ¶ИТӘЗуёЯЎЈДҝЗ°,ЦчТӘөДDIA ·Ҫ·ЁөДЩ|ЧV’ЯГиЛЩ¶ИҫщФЪ10 Hz ЧуУТ,АэИзSWATH өИ,ТтҙЛІЙУГЭ^ҙуЯx“сҙ°ҝЪ(25 ~50 amu),ТФҝs¶МСӯӯh(huЁўn)•rйg(јs2. 5 ~3. 2 s)ЎЈQ-qIT-OT ИэәПТ»Щ|ЧVјҜЛДҳO—UЎўҫҖРФлxЧУЪеәНOrbitrap 3 ·NЩ|Бҝ·ЦОцЖчһйТ»уw,Orbitrap ’ЯГиЛЩ¶ИЯ_өҪ15 Hz,ҫҖРФлxЧУЪеЯ_өҪ20 Hz,һйҝs¶МӮчҪy(tЁҜng)DIA өДЯx“сҙ°ҝЪЎўҪөөНёЙ”_ЎўМбёЯм`Гф¶ИМṩБЛҝЙДЬ[10] ЎЈ
ұҫСРҫҝКЧПИ»щУЪЛДҳO—UәНOrbitrap,ҪЁБўБЛҪӣөдDIA ·Ҫ·Ё(ҲD1A):К№УГ20 amu Яx“сҙ°ҝЪ,АыУГOrbitrap 30K ёЯ·ЦұжВКәНHCD ёЯДЬЛйБС,ТАҙОЯx“сЎўЛйБСЎў’ЯГиХыӮҖЩ|Бҝ·¶Үъ(m / z 500-900),Сӯӯh(huЁўn)•rйgғHһй1. 9 s,”ө“юМҺАн•rМфЯxҸҠ¶ИЧоёЯөДЧУлxЧУҪMіЙДёЧУлxЧУҢҰЯMРРЙ«ЧV·еМбИЎәН¶ЁБҝЎЈҙЛНв,АыУГҫҖРФлxЧУЪеөДёЯм`Гф¶ИәНёЯ’ЯГиЛЩ¶И,ҪЁБўБЛғЙ·N»щУЪТ»јү¶ЁБҝөДРВ·Ҫ·Ё:Ң’ҙ°ҝЪSIM ’ЯГиDIA (WiSIM-DIA)әНИ«’ЯГиDIA (Full MS-DIA)ЎЈ
WiSIM-DIA(ҲD1B)АыУГ200 amu Ң’ҙ°ҝЪәYЯxДёлxЧУ,ФЪm / z 400 ~1000 ·¶ҮъТАҙОК№УГOrbitrap ЯMРР240 K і¬ёЯ·ЦұжлxЧУұO(jiЁЎn)ңy’ЯГи,ЧоҙуіМ¶ИЕЕіэ»щЩ|ёЙ”_,»щУЪТ»јүҫ«ҙ_Щ|Бҝ”өЯMРР¶ЁБҝЎЈФЪГҝӮҖSIM’ЯГиЦ®әу,АыУГ12 amu Хӯҙ°ҝЪЯx“сДёлxЧУ,ФЪЩ|Бҝ·¶ҮъғИТАҙОЯMРРҫҖРФлxЧУЪе¶юјүЛйЖ¬лxЧУ’ЯГи,»щУЪ¶юјүЧVҲDҢҚ¬F(xiЁӨn)¶ЁРФҙ_ЧCЎЈ
Full MS-DIA(ҲD1C)ФЪWiSIM-DIA »щөAЙПҢў12 amu Яx“сҙ°ҝЪҝs¶МөҪ3 amu,Я_өҪЕcDDA Па®”өДЯx“сҙ°ҝЪ,ө«’ЯГиҙ°ҝЪ”өБҝФцјУБЛ4 ұ¶ЎЈһйБЛұЬГвСӯӯh(huЁўn)•rйgЯ^йLҢҰ¶ЁБҝФміЙөДУ°н‘,ФЪm / z 400 ~1000 өДЩ|Бҝ·¶ҮъғИІеИл5 ӮҖOrbitrap 240 K і¬ёЯ·ЦұжТ»јүИ«’ЯГиТФұЈЧC’ЯГиьc”өЎЈFull MS-DIA Т»јү’ЯГийgёфғH2. 6 s,ұЈЧCБЛ¶ЁБҝҪY№ыөДҝЙҝҝРФЎЈ
лД¶Ој{БчТәПа·ЦлxөДіц·е•rйgТ»°гФЪ30 s ТФЙП,ТтҙЛФЪәПЯmөДСӯӯh(huЁўn)•rйgғИ,3 ·NDIA ·Ҫ·ЁҝЙТФёщ“юҳУұҫҸНлsіМ¶Им`»оХ{№қ(jiЁҰ)Щ|Бҝ·¶ҮъЎў·ЦұжВКЎўЯx“сҙ°ҝЪөИ…ў”ө,Я_өҪЧојС·ЦОцР§№ыЎЈАэИз,·ЦОц·ЦЧУБҝЭ^ҙуөДлД¶О•r,Щ|Бҝ·¶ҮъҝЙТФХ{Хыһйm / z 400 ~1200 »тёьҢ’ЎЈ
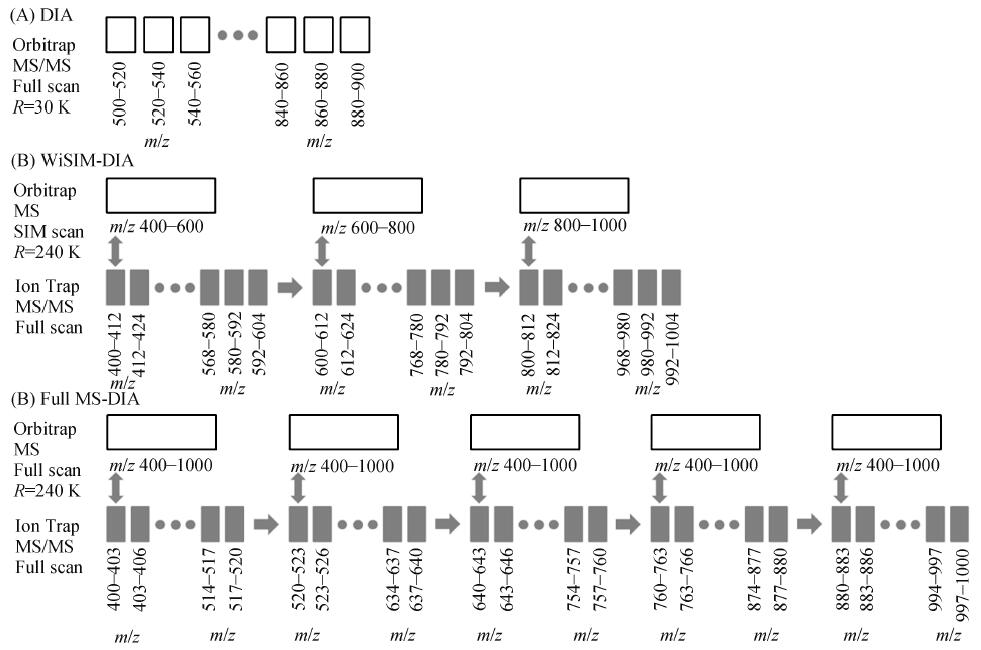
ҲD1 »щУЪQ-qIT-OT өД3 ·N”ө“ю·ЗТАЩҮРФІЙјҜФӯАнЎЈ(A)Ҫӣөд”ө“ю·ЗТАЩҮРФІЙјҜDIA;(B)Ң’ҙ°ҝЪSIM ’ЯГи
DIA (WiSIM-DIA);(C)И«’ЯГиDIA (Full MS-DIA)
Fig. 1 Schematic illustrating quadrupole-ion-orbitrap (Q-qIT-OT) based three data-independent acquisition (DIA) methods. (A) Classic DIA, (B) Wide isolation window SIM scan DIA (WiSIM-DIA), (C) Full scan DIA (Full MS-DIA)
3.2 ҸНлs»щЩ|јУҳЛҳУұҫөД”ө“юІЙјҜЕc·ЦОц
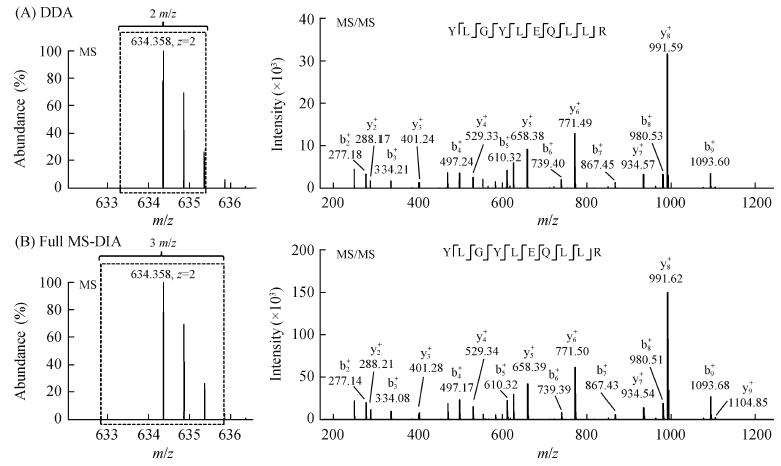
һйБЛҝјІмЕcұИЭ^3 ·NDIA ·Ҫ·ЁөД·ЦОцР§№ы,ҢҚтһҢҰHela јҡ°ыИ«ө°°ЧГёҪвТәЦРМнјУөД10 —lөНқв¶ИлД¶ОЯMРРБЛ”ө“юІЙјҜЎЈ10 —lәПіЙөДҳЛңКлД¶ОҫщҒнЧФУЪ·ЗИЛФҙөДЦІОпө°°Ч,·ЦЧУБҝФЪ1100 ~1700 Da Ц®йg,ҫЯУРёЯ¶ИМШ®җРФЎЈҳЛңКлД¶ОИЬТәК№УГ100 mg/ L Hela јҡ°ыГёҪвТәЦрјүПЎбҢһй7 ·Nқв¶ИМЭ¶И,ІўЦрТ»К№УГ3 ·NDIA ·Ҫ·ЁІЙјҜ(ЙПҳУБҝ1 ЧМL),ҝјІм·Ҫ·ЁөДЯx“сРФЎўҫҖРФЎўЦШ¬F(xiЁӨn)РФәНм`Гф¶ИЎЈҲD2 Х№КҫБЛ50 mg/ L қв¶ИлД¶ОҳУұҫөДҝӮлxЧУБчҲD(TIC)ЎўДҝҳЛлД¶ОYLGYLEQLLR (m / z 634. 356, 2+)өДМбИЎлxЧУБчҲD(XIC)әНЧУлxЧУЧVҲD(MS/ MS)ЎЈУЙУЪHela јҡ°ыҳУұҫёЯ¶ИҸНлs,ҳЛңКлД¶ОөДЙ«ЧV·еФЪTIC ЦРұ»НкИ«СЪЙwЎЈБнНв,ПаұИWiSIM-DIA әНFull MS-DIA,DIA Ц»УР¶юјү’ЯГиӣ]УРТ»јү’ЯГи,ТтҙЛTIC ҫщУЙ¶юјүЧVҲDҳӢіЙ,РЕФлұИПаҢҰЭ^өНЎЈЯMТ»ІҪМбИЎФ“лД¶ОөДXIC ҲD,ЖдЦРDIA »щУЪЧУлxЧУ¶ЁБҝ,Яx“сЧоҸҠЧУлxЧУm / z 991. 557 РОіЙ634. 356 ~991. 557 ДёЧУлxЧУҢҰМбИЎЙ«ЧV·е;WiSIM-DIA әНFull MS-DIA»щУЪДёлxЧУ¶ЁБҝ,АыУГДёлxЧУҫ«ҙ_Щ|Бҝ”өm / z 634. 356 МбИЎЙ«ЧV·еЎЈНЁЯ^ұИЭ^XIC ҝЙТФҝҙіц,ФЪҸНлsHela јҡ°ы»щЩ|ЦР,3 ·N·Ҫ·ЁҫщДЬУРР§ЕЕіэёЙ”_ЎЈ
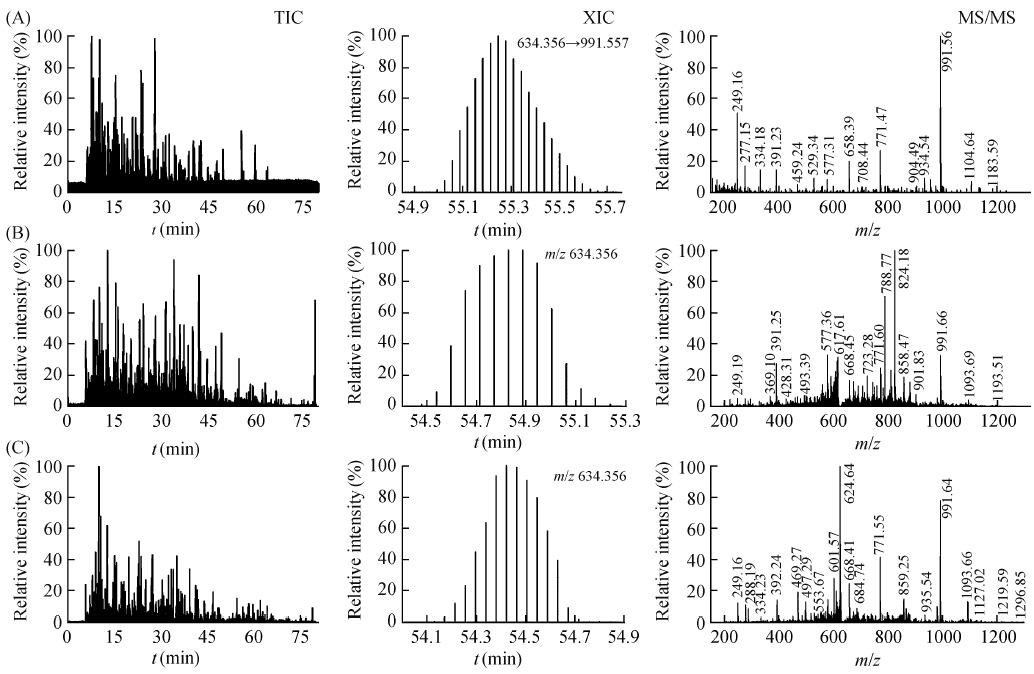
ҲD2 3·N”ө“ю·ЗТАЩҮРФІЙјҜ·ЦОцHela јҡ°ыҳУұҫЦРөДҳЛңКлД¶ОЎЈ(A) DIAЎў(B) WiSIM-DIA әН(C) Full MS-DIA өДTIC ҲDЎўлД¶ОYLGYLEQLLR лxЧУҢҰXIC ҲDәНMS/ MS ҲD
Fig. 2Analysis of standard peptides spiked in Hela cell digest by the three DIA methods. TICs, peptide YLGYLEQLLR XICs and MS/ MS spectra of (A) classic DIA, (B) WiSIM-DIA and (C) Full MS-DIA data
лД¶ОYLGYLEQLLR өДіц·е•rйgјsһй35 s,3 ·NDIA ФЪіц·е•rйgғИ’ЯГиьc”өҫщЯ_өҪ10 ТФЙП,·Ц„eһй22, 14, 14,ҫЯУРБјәГөДЙ«ЧV·еРНәНҝЙҝҝөД¶ЁБҝ·еГж·eЎЈ
MS/ MS ЧVҲDп@КҫБЛ3 ·NЯx“сҙ°ҝЪПВ¶юјүЛйЖ¬лxЧУөД’ЯГиҪY№ыЎЈDIA өДЯx“сҙ°ҝЪЧоҙу(20 amu),Щ|ЧV·еЧо¶а,ө«Orbitrap ёЯ·Цұж’ЯГиДЬүтУРР§…^(qЁұ)·ЦЛйЖ¬лxЧУ,ТтҙЛЧVҲDҫЯУРБјәГөДРЕФлұИ,ҝЙТФУРР§УГУЪ¶ЁРФЕc¶ЁБҝ·ЦОцЎЈWiSIM-DIA ҙ°ҝЪҝs¶Мһй12 amu,ө«ҢЩУЪҫҖРФлxЧУЪеөН·Цұж’ЯГи,ТтҙЛЩ|ЧV·еөД…^(qЁұ)·ЦДЬБҰПаҢҰЭ^Их,ФміЙТ»¶ЁёЙ”_ЎЈFull MS-DIA ҙ°ҝЪғH3 amu,ЕcDDA Па®”,°С»щЩ|ёЙ”_ҪөөҪБЛЧоөН,МṩБЛБјәГөД¶ЁРФҙ_ЧCРЕПўЎЈ
3.3 ·Ҫ·ЁөДҫҖРФЎўм`Гф¶ИөДҝјІмЕcұИЭ^
”ө“юЯMТ»ІҪК№УГPinpoint 1. 4 ЬӣјюМҺАн,ҝјІм·Ҫ·ЁФЪ100 ng ҸНлsHela јҡ°ы»щЩ|ПВ,20 ~200 pg өНқв¶ИлД¶О·¶ҮъғИөДҫҖРФЎўЦШ¬F(xiЁӨn)РФәНм`Гф¶ИЎЈІўТФ10 —lҳЛңКлД¶ОөДPRM ”ө“юиb¶ЁҪY№ыЧчһйЧVҲDҺм,ЯMРРЧVҲDЖҘЕдәН¶ЁРФҙ_ЧCЎЈDIA ЯxИЎҳЛңКЧVҲDЦРҸҠ¶ИЧоёЯөД8 ӮҖЧУлxЧУЧчһй¶ЁРФҙ_ЧCТА“ю,ІўМбИЎЖдЦРЧоәГөД2 ӮҖy лxЧУЯMРР¶ЁБҝ·ЦОцЎЈWiSIM-DIA әНFull MS-DIA Н¬ҳУЯxИЎЧоҸҠөД8 ӮҖЧУлxЧУЯMРР¶ЁРФҙ_ЧC,¶ЁБҝ·ЦОц„tК№УГТ»јүДёлxЧУөДҶОН¬О»ЛШ·еәНПааҸөДөЪТ»ӮҖН¬О»ЛШ·еЎЈ
ТФлД¶ОYLGYLEQLLR ( m / z 634. 356, 2+) һйАэ, DIA ”ө“юЯxИЎЧоҸҠөДy8 ( m / z 991. 557) әНy6 (m / z 771. 472)Ччһй¶ЁБҝлxЧУҢҰ,АLЦЖҳЛңКЗъҫҖ(ҲD3A)ЎЈЗъҫҖФЪ100 fg ~ 50 pg ·¶ҮъғИп@КҫіцБјәГөДҫҖРФ,Н¬•r, 8 ӮҖ¶ЁРФҙ_ЧCлxЧУЕcЧVҲDҺмЦРөДҳЛңКЧVҲDёЯ¶ИЖҘЕд,ҸҠ¶ИұИАэТ»ЦВЎЈWiSIM-DIA ЯxИЎҶОН¬О»ЛШ·еM (m / z 634. 356)әНПааҸН¬О»ЛШ·еM+1 (m / z 634. 857)ЯMРР¶ЁБҝ·ЦОц,ФЪ100 fg ~200 pg ·¶ҮъғИҫЯУРБјәГөДҫҖРФкPПө(ҲD3B)ЎЈWiSIM-DIA ¶юјүһйөН·Цұж’ЯГи,ТЧКЬ»щЩ|әН№ІБчіцлД¶ОЛйЖ¬өДёЙ”_,ө«УЙУЪЯx“сҙ°ҝЪЭ^РЎ,ТтҙЛТІп@КҫіцБјәГөДЧУлxЧУЧVҲDЖҘЕдҪY№ыЎЈFull MS-DIA ¶ЁБҝБчіМЕcWiSIM-DIAНкИ«Т»ЦВ,·Ҫ·ЁФЪ500 fg ~200 pg ·¶ҮъғИп@КҫіцБјәГөДҫҖРФкPПө,Н¬•r¶ЁРФҙ_ЧCҪY№ыБјәГ(ҲD3C)ЎЈ
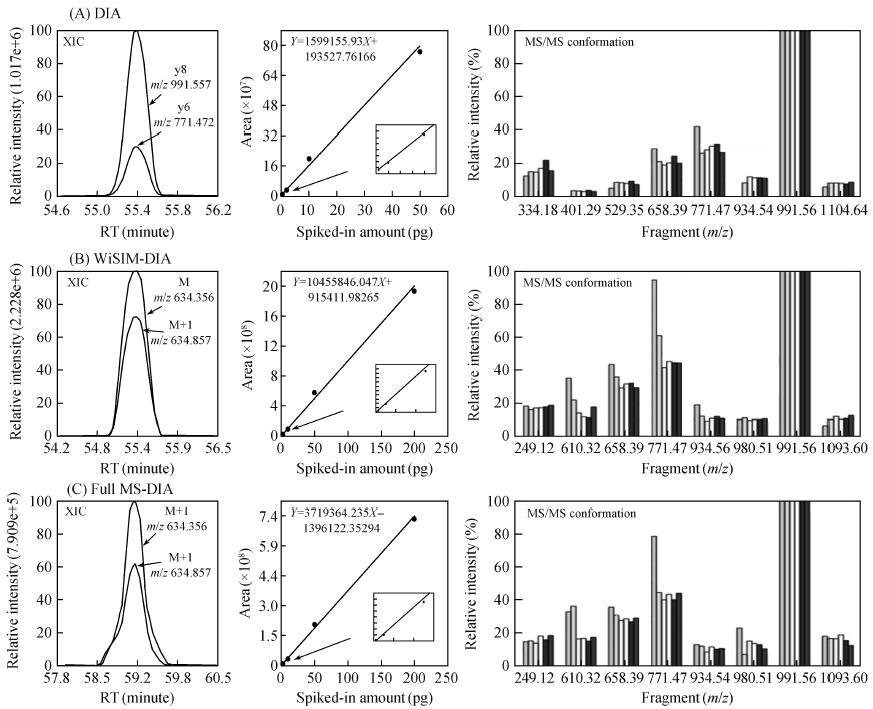
ҲD3 лД¶ОYLGYLEQLLR К№УГ3 ·N”ө“ю·ЗТАЩҮРФІЙјҜөД¶ЁРФ¶ЁБҝҪY№ыЎЈ(A) DIAЎў(B) WiSIM-DIA әН(C) Full MS-DIA өД¶ЁБҝлxЧУҢҰXIC ҲDЎўҳЛңКЗъҫҖәНЧVҲDЖҘЕдҙ_ЧCҲDЎЈЖҘЕдҙ_ЧCЦщ оҲDҸДЧуөҪУТТАҙОһй0. 5, 2, 10, 50 әН200 mg/ L әНҳЛңКЧVҲD
Fig. 3 Confirmation and quantification of peptide YLGYLEQLLR using the three DIA methods. Quantification ion XICs, standard curves and spectra library matching charts of (A) classic DIA, (B) WiSIM-DIA and (C) Full MS-DIA data. The columns of spectra library matching charts represent 0. 5, 2, 10, 50 and 200 mg/ L and library spectrum from left to right, respectively
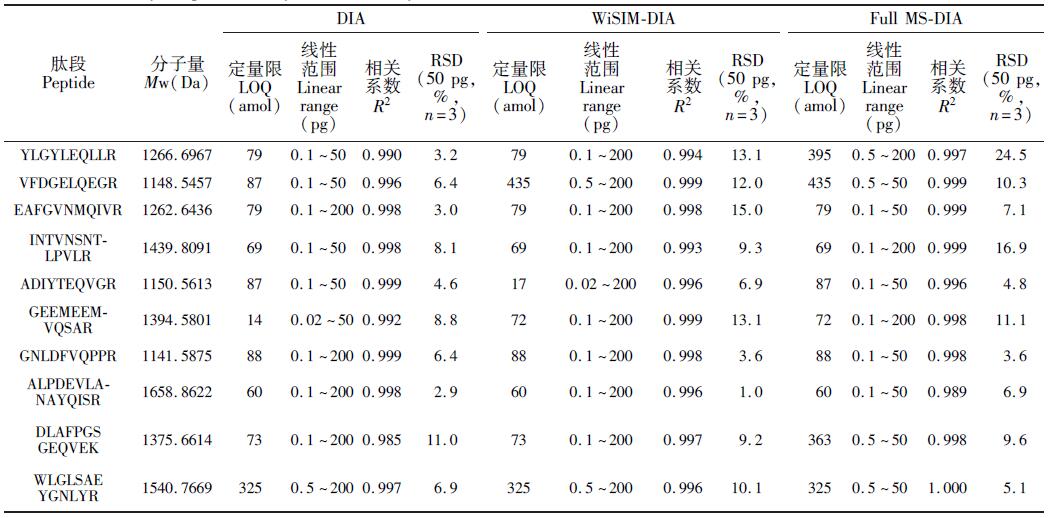
ҢҰ10 —lлД¶ОөД·ЦОцҪY№ы…RҝӮ(ұн1),ҪY№ыұнГч,3 ·NDIA ·Ҫ·ЁФЪfg ~ pg ”өБҝјүлД¶О·¶ҮъғИҫщп@КҫіцБјәГөДҫҖРФЎўЦШ¬F(xiЁӨn)РФәНм`Гф¶ИЎЈ3 ·N·Ҫ·ЁөДЧоөН¶ЁБҝПЮҫщЯ_өҪamol јү,ЖдЦРлД¶ОGEEMEEMVQSARФЪDIA ЦРЧоөН¶ЁБҝПЮЯ_14 amol,і¬ФҪБЛіЈТҺ(guЁ©)SRM әНPRM өД¶ЁБҝЛ®ЖҪЎЈұИЭ^3 ·N·Ҫ·ЁҝЙТФҝҙіц,DIAЕcWiSIM-DIA ҪY№ыІо®җІ»ҙу,ЧCГчБЛ»щУЪёЯ·ЦұжөД¶юјү¶ЁБҝәН»щУЪі¬ёЯ·ЦұжөДТ»јү¶ЁБҝЯx“сРФПа®”,ҫщДЬУРР§ЕЕіэ»щЩ|әН№ІБчіцлД¶ОөДёЙ”_,¶ЁБҝДЬБҰіцЙ«ЎЈғЙ·N·Ҫ·ЁУЦёчУРМШьc,РОіЙғһ(yЁӯu)„Э»ҘСa:DIA НЁЯ^ЛДҳO—UәНOrbitrap ғЙјүәYЯx,ДЬУРР§·ЦОцҳOҸНлsөДҳУЖ·;WiSIM-DIA К№УГДёлxЧУ¶ЁБҝ,ұЬГвБЛЛйБСЯ^іМЦРөД“pК§,ФЪПаҢҰәҶҶОөД»щЩ|ЦРм`Гф¶ИёьёЯЎЈ
ұн1 3·N”ө“ю·ЗТАЩҮРФІЙјҜөДҫҖРФ·¶ҮъЎўЦШ¬F(xiЁӨn)РФЎўм`Гф¶И
Table 1 Linearity, reproducibility and sensitivity of the three DIA methods
3. 4 ЦұҪУЛСҺмиb¶ЁөДҝјІмЕcұИЭ^
УЙУЪЯx“сҙ°ҝЪЯ^ҙу,Н¬•r¶юјүЧVҲDҹo·ЁЕcТ»јүДёлxЧУПакPВ“(liЁўn),ӮчҪy(tЁҜng)”ө“ю·ЗТАЩҮРФІЙјҜҹo·ЁЦұҪУЛСҺм,РиТӘЧVҲDҺмЖҘЕдІЕДЬЯMРР¶ЁРФҙ_ЧC,К№DIA өД‘ӘУГКЬЦЖУЪDDAЎЈ
Full MS-DIA »щУЪТ»јүИ«’ЯГи¶ЁБҝ,ДёлxЧУОҙҪӣЯ^З°јүЩ|Бҝ·ЦОцЖчЯx“с,ТтҙЛПаұИDIA әНWiSIM-DIA КЬөҪөДёЙ”_ёьҙу,м`Гф¶ИВФөНЎЈө«КЗ,Full MS-DIA Ңў¶юјүЯx“сҙ°ҝЪҝs¶МөҪ3 amu,ЕcӮчҪy(tЁҜng)DDA өДЯx“сҙ°ҝЪПа®”,ДЬүтЧчһйөН·Цұж”ө“ю,ЦұҪУУГУЪ”ө“юҺмҷzЛч(Па®”УЪДёлxЧУЩ|Бҝҫ«¶ИһйТА1. 5 amu),”[Г“БЛЧVҲDҺмөДПЮЦЖ,ҢҚ¬F(xiЁӨn)БЛDDA ЕcDIA өДҪy(tЁҜng)Т»ЎЈ
ҲD4 Х№КҫБЛлД¶ОYLGYLEQLLR ЧVҲDөДЦұҪУЛСҺмҪY№ыЎЈDDA НЁіЈТФ2 amu һйЯx“сҙ°ҝЪ,ЕcFull MS-DIA 3 amu Яx“сҙ°ҝЪПаІоІ»ҙуЎЈЛСҺм•r,Full MS-DIA Т»јүЩ|Бҝҫ«¶ИТФҙ°ҝЪҢ’¶ИһйПЮ,јҙТА1. 5 amu,оҗЛЖУЪөН·ЦұжЩ|ЧVDDA ”ө“юөДЛСҺмиb¶ЁЎЈҪY№ып@Кҫ,Full MS-DIA ЕcDDA өД¶юјүЧVҲDёЯ¶ИПаЛЖ,лmИ»Full MS-DIA ЧVҲDөИН¬УЪөН·Цұж”ө“ю,ө«иb¶ЁҪY№ыӣ]УРГчп@Іо®җ,РтБРЖҘЕдНкИ«Т»ЦВЎЈ
ҲD4лД¶ОYLGYLEQLLR ЧVҲDЕcЛСҺмиb¶ЁҪY№ыЎЈ(A) DDA әН(B) Full MS-DIA өДТ»јүЯx“сҙ°ҝЪәН¶юјүиb
¶ЁҲD
Fig. 4Spectra and database search results of peptide YLGYLEQLLR. MS isolation window and MS/ MS identifica-tion of (A) data dependent acquisition (DDA) and (B) Full MS-DIA data
һйБЛҝјІмFull MS-DIA УГУЪЛСҺмиb¶ЁөДҝЙРРРФ,ҢҚтһЯMТ»ІҪҢҰ100 ng Hela јҡ°ыИ«ө°°ЧЯMРРDDA әНFull MS-DIA ІЙјҜ,”ө“юК№УГUniprot ИЛоҗө°°Ч”ө“юҺмЛСҺмиb¶Ё(ұн2)ЎЈЖдЦР,DDAЎўFull MS-DIA Т»јүЩ|Бҝҫ«¶И·Ц„eһй10 ppmЎў1. 5 Da,¶юјүЩ|Бҝҫ«¶Иҫщһй0. 6 Da,ҝЁЦөҳЛңКһйq<0. 01 (Percolator)ЎЈҪY№ып@Кҫ,DDA №Іиb¶Ё15575 —lлД¶О,ҢҰ‘Ә3228 ӮҖ·ЗИЯУаө°°Ч;Full MS-DIA №Іиb¶Ё10515 —lлД¶О,ҢҰ‘Ә2835 ӮҖ·ЗИЯУаө°°ЧЎЈәуХЯиb¶ЁҪY№ыЙЩУЪЗ°ХЯ,ЦчТӘКЗғЙХЯ’ЯГи·¶ҮъІ»Н¬ФміЙөД,Full MS-DIA Риҝј‘]’ЯГиьc”ө,ТтҙЛ’ЯГи·¶ҮъЭ^Хӯ;ҙЛНв,ғЙХЯө°°Ч”өөДІо®җГчп@РЎУЪлД¶О”ө,Я@КЗУЙУЪ·ЗИЯУалД¶ОЦчТӘјҜЦРФЪm / z 400 ~1000 ·¶Үъ,’ЯГи·¶ҮъЭ^ХӯҢҰө°°Чиb¶ЁУ°н‘Э^РЎЎЈ
ұн2 100 ng Hela јҡ°ыИ«ө°°ЧҳУұҫDDA ЕcFull MS-DIA ’ЯГи…ў”өЎўЛСҺм…ў”өәНиb¶ЁҪY№ы(n =3)
Table 2 MS scan parameters, database search parameters and identification results of DDA and full MS-DIA analysis of 100 ng Hela cell tryptic digest (n =3)

DDA ЕcFull MS-DIA өДө°°Чиb¶ЁҪY№ыЦШәП¶ИёЯ,ұнГчЩ|Бҝҫ«¶ИҢҰиb¶ЁҪY№ыУ°н‘І»ҙу(ҲD5A)ЎЈНЁЯ^ұИЭ^лД¶ОVVIGMDVAASEFFR өД¶юјүЧVҲDЖҘЕдҝЙТФҝҙіц,ғЙ·N·Ҫ·ЁІЙјҜөД¶юјүЧVҲD·ЗіЈПаЛЖ,ЛйЖ¬ЖҘЕдТІҺЧәхТ»ЦВ(ҲD5B)ЎЈТФЙП”ө“юЧCГч,Full MS-DIA ҝЙТФЦұҪУУГУЪ”ө“юҺмҷzЛчиb¶Ёө°°Ч,Іў«@өГҝЙҝҝөДиb¶ЁҪY№ы,ҢҚ¬F(xiЁӨn)БЛDDA ЕcDIA өДҪy(tЁҜng)Т»ЎЈ
ҲD5 100 ng Hela јҡ°ыИ«ө°°ЧҳУұҫDDA ЕcFull MS-DIA иb¶ЁҪY№ыұИЭ^ЎЈ(A) ғЙ·N·Ҫ·Ёө°°Чиb¶ЁҪY№ыұИЭ^;(B) ғЙ·N·Ҫ·Ё¶юјүЧVҲDЖҘЕдұИЭ^(лД¶ОVVIGMDVAASEFFR)
Fig. 5 Comparison of identification results of 100 ng Hela cell digest by DDA and Full MS-DIA methods. (A) Comparison of identified number of proteins, (B) Comparison of identified MS/ MS spectra (peptide VVIGMD-VAASEFFR)

4ҪYХ“
»щУЪмoлҠҲцЬүөАЪеQ-qIT-OT Щ|ЧVҪЁБўDIAЎўWiSIM-DIAЎўFull MS-DIA 3 ·N”ө“ю·ЗТАЩҮРФІЙјҜ·Ҫ·Ё, ІўК№УГМнјУ10 —lөНқв¶ИлД¶ОөДHela јҡ°ыИ«ө°°ЧҳУұҫҢҰ·Ҫ·ЁЯMРРҝјІмЎЈҪY№ыұнГч,3 ·N·Ҫ·ЁөД¶ЁБҝПЮҫщФЪ14 ~435 amol ·¶ҮъғИ,ҫҖРФкPПөЕcЦШ¬F(xiЁӨn)РФБјәГ,¶ЁРФҙ_ЧCҝЙҝҝРФёЯЎЈЖдЦР,WiSIM-DIA »щУЪі¬ёЯ·ЦұжSIM ¶ЁБҝ,ЕcҪӣөдDIA ¶юјү¶ЁБҝҫЯУРТ»¶ЁөД»ҘСaРФ;¶шFull MS-DIA Ңў¶юјүЯx“сҙ°ҝЪҝs¶МөҪ3 amu,ҢҚ¬F(xiЁӨn)БЛDIA ”ө“юЦұҪУЛСҺмиb¶Ё,№ІҸД100 ng Hela јҡ°ыИ«ө°°ЧҳУұҫиb¶ЁөҪ2835 ӮҖ·ЗИЯУаө°°Ч,ЕcDDA иb¶ЁҪY№ыЦШәП¶ИёЯЎЈ»щУЪQ-qIT-OT өД„“(chuЁӨng)РВ”ө“ю·ЗТАЩҮРФІЙјҜ·Ҫ·Ёһй¶ЁБҝө°°ЧЩ|ҪMҢWМṩБЛИ«РВТ•ҪЗЕcІЯВФЎЈ
References
1 Kalli A, Smith G T, Sweredoski M J, Hess S. J. Proteome Res. , 2013, 12(7): 3071-3086
2 Nagaraj N, Kulak N A, Cox J, Neuhauser N, Mayr K, Hoerning O, Vorm O, Mann M. Mol. Cell. Proteomics, 2012,11(3): M111. 013722
3 Geiger T, Wehner A, Schaab C, Cox J, Mann M. Mol. Cell. Proteomics, 2012, 11(3): M111. 014050
4 Gallien S, Duriez E, Crone C, Kellmann M, Moehring T, Domon B. Mol. Cell. Proteomics, 2012, 11(12): 1709-1723
5 Gallien S, Duriez E, Demeure K, Domon B. J. Proteomics, 2013, 81: 148-158
6 Chapman J D, Goodlett D R, Masselon C D. Mass Spectrom. Rev. , 2013: 10. 1002/ mas. 21400
7 Law K P, Lim Y P. Expert Rev. Proteomics, 2013, 10(6): 551-566
8 Gillet L C, Navarro P, Tate S, Rost H, Selevsek N, Reiter L, Bonner R, Aebersold R. Mol. Cell. Proteomics, 2012,11(6): O111. 016717
9 Egertson J D, Kuehn A, Merrihew G E, Bateman N W, MacLean B X, Ting Y S, Canterbury J D, Marsh D M, Kellmann M, Zabrouskov V, Wu C C, MacCoss M J. Nat. Methods, 2013, 10(8): 744-746
10 Senko M W, Remes P M, Canterbury J D, Mathur R, Song Q, Eliuk S M, Mullen C, Earley L, Hardman M, Blethrow J D, Bui H, Specht A, Lange O, Denisov E, Makarov A, Horning S, Zabrouskov V. Anal. Chem. , 2013, 85(24):11710-11714
Quantification Analysis of Targeted Proteins in Complex Sample by Novel Data Independent Acquisition
ZHANG Wei1 , Reiko Kiyonami2 , JIANG Zheng*1 , CHEN Wei1
1(Thermo Fisher Scientific, Shanghai 201206, China)
2(Thermo Fisher Scientific, San Jose, CA, USA)
AbstractData independent acquisition ( DIA) is a novel MS scan mode for quantitative proteomics,acquiring all precursors as well as fragments without any loss of low abundant ions, and breaks the throughput limitation of product ion quantification by high-resolution MS. Here we developed three DIA methods on quadrupole-linear ion trap-Orbitrap (Q-qIT-OT) Tribrid MS, classic DIA, as well as novel wide isolation window SIM scan (WiSIM)-DIA and full scan-DIA (Full MS-DIA). Quantitative analysis of 10 low abundant peptides spiked in Hela cell digest was performed by the three methods for linearity, reproducibility and sensitivity evaluation. The results showed that the LOQs reached amol level (14 -435 amol) with good linearity and effective MS/ MS confirmation. WiSIM-DIA utilizes ultra-high resolution SIM scan for quantification complementary with classic DIA. The isolation window of Full MS-DIA was down to 3 amu, and the data could be directly used for database searching, thus realizing the integration of data dependent acquisition (DDA) and DIA, and avoiding the limitation of using spectra library.
Keywords Orbitrap; Data independent acquisition; Proteomics; Absolute quantification
(Received 24 April 2014; accepted 4 July 2014Ј©
°l(fЁЎ)ІјХЯЈәЩҗД¬пwКА –ҝЖјјЈЁЙ«ЧVЕcЩ|ЧVЈ©
В“(liЁўn)ПөлҠФ’Јә13386161207
E-mailЈәping.shen2@thermofisher.com
В“(liЁўn)ПөлҠФ’Јә13386161207
E-mailЈәping.shen2@thermofisher.com

- ёЯ·ЦұжВКҝХйgЩ|ЧVө°°ЧЩ|ҪMҢWЖҪЕ_PLATOөДјјРgғһ(yЁӯu)„ЭЕc‘ӘУГ
- TGA-FTIR-GCMSВ“(liЁўn)УГПөҪy(tЁҜng)ФЪ¶аоIУт‘ӘУГ°ёАэ·ЦПн
- ЩҗД¬пwҶО—UЩ|ЧVУГУЪјҸҝ—Ж·ЦРөДAPәНAPnEOҷzңy
- лxЧУЙ«ЧV-ёЯ·ЦұжЩ|ЧVВ“(liЁўn)УГјјРgУГУЪ·ЦОцСӘТәЦРҸҠҳOРФ¶ҫОп
- ЦШьcИХУГПыЩMЖ·УРәҰ»ҜҢWОпЩ|ёЯ·ЦұжЩ|ЧVЧVҺмөДҳӢҪЁј°‘ӘУГ
- iCAP MSX ICP-MSЦъБҰКҜУН»Ҝ№ӨРРҳI(yЁЁ)І»Н¬·РіМөДөдРНҳУЖ·іЈТҺ(guЁ©)·ЦОц
- spICP-TOF-MSјјРgУГУЪИјГәФҙҙЕиFөVҶОј{ГЧоwБЈ¶акPжIЦВ¶ҫҪM·ЦХз„e
- ФЪҫҖCI-OrbitrapјјРgЦъБҰҙуҡвЦРҡв‘B(tЁӨi)ә¬СхУРҷC·ЦЧУСРҫҝИЎөГРВЯMХ№
- ЩҗД¬пw”yCNHUPOіЙ№ҰЕeЮkҪMҢWЗ°СШ„“(chuЁӨng)РВјјРgёЯ·еХ“үҜ
- CPHIЙъГьҝЖҢWЕcЙъОпбt(yЁ©)ЛҺЦчо}В“(liЁўn)Х№јҙҢўЦШ°хҶўД»
- CISILE 2025ҝЖҢWғxЖчј°ҢҚтһКТХ№ХРЙМјҙҢўҲAқMКХ№Щ
- Analytica 2026өВҮшДҪДбәЪ·ЦОцЙъ»ҜІ©У[•юНЁЦӘ
- ЩҗД¬пwКЧЕ_Үш®aТәПаЙ«ЧVёЯ·ЦұжЩ|ЧVВ“(liЁўn)УГғxХэКҪПВҫҖ
- SCIEXИ«ЗтёұҝӮІГІМҝЎЛЙ:®”Щ|ЧVҫЮо^ӣQ¶ЁНШХ№ФЪИAЙъ®a
- °ІҪЭӮҗЕcәП·КҫCәПРФҮшјТҝЖҢWЦРРДӯh(huЁўn)ҫіСРҫҝФә
- °ІҪЭӮҗХ№КҫИ«РВInfinity IIIІўёьРВ‘р(zhЁӨn)ВФЯMХ№


Copyright(C) 1998-2025 ЙъОпЖчІДҫW лҠФ’Јә021-64166852;13621656896 E-mailЈәinfo@bio-equip.com